Writing Documentation in XML
- Scott Saccone
Documentation on the genomic relational databases in BioQ can be written in XML. The XML files must be processed by the dbdoc_util.pl program which saves the documentation in a relational database. This document describes the format used by the XML file. Note that dbdoc_util.pl has an option dbdoc-target-db which specifies which BioQ genomic database is being documented, so that all the documentation below is relative to this option.
Note: a number of new elements have been added to the XML schema which are not yet documented. These pertainly mainly to genomic features, and include these elements:
- feature_keywords
- feature_tables
- db_feat_tables
- feature_table_population
- genomes
- groups
Examples of these elements can be found on our Subversion server in the files features.xml and bioq_ensembl_hs_core_64_37.xml.
Contents
Examples
We are working on moving this source code to a public machine so that these XML files can be viewed by anyone.
Example | Description |
|---|---|
bioq_hapmap_core.xml | Core HapMap documentation - the bulk of the documentation that should not change by much between different releases of the database. |
bioq_hapmap_p3r3_dbsnp132.xml | Documentation specific to this release of the database. |
bioq_hapmap_pr28_dbsnp132.xml | Documentation specific to this release of the database. |
Overview
All documentation is contained in the <documentation> root element. The next level of elements include:
Element | Description |
|---|---|
<db> | A database |
<tags> | Divides tables into logical categories |
<tables> | The tables in a genomic database |
<flow_groups> | Flow groups in the BERT model |
<processes> | Processes in the BERT model |
<queries> | SQL queries used in BioQ query page |
Databases: the <db> element
Specify documentation for a single BioQ genomic database.
Example
<documentation>
<db>
<label>HapMap Phase III R3</label>
<version>Phase III Release 3</version>
<default_filter>ASW</default_filter>
<short_descrip>Data from Phase 3 Release 3 of the international <a href="http://hapmap.ncbi.nlm.nih.gov" target="_blank">HapMap</a> project</short_descrip>
<long_descrip>
<p>This database contains genotyping, allele frequency and linkage disequilibrium (LD) data from
Phase III Release 3 of international HapMap project.
Physical mapping data from <a href="http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp&cmd=search&term=" target="_blank">dbSNP</a>
build 132 (BioQ: _db_link(dbsnp_human_132)) was used to determine genomic intervals for estimating LD.
</p>
</long_descrip>
</db>
</documentation>
This leads to the following documentation on the BioQ main page.
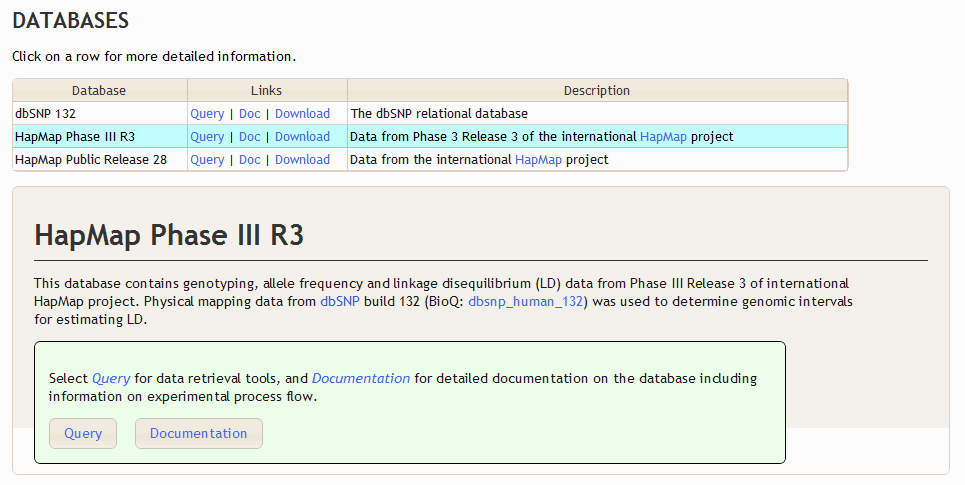
Child elements of <db>
Element | Description | Notes |
|---|---|---|
<label> | Label used for the database in BioQ |
|
<default_filter> | Default filter | Only when filters exist |
<long_descrip> | Long description |
|
Categories of tables: the <tags> element
This classifies tables into categories. The categories are shown in the database page.
Example
<documentation>
<tags>
<tag tag_name="Population \_uc_($r)" repeat="asw ceu chb chd gih jpt lwk mex mkk tsi yri">
<short_description>Tables related to the \_uc_($r) population</short_description>
<tables>/^$r_/</tables>
</tag>
</tags>
</documentation>
Which results in the "Population" entries below in the Categories section of the BioQ HapMap database page.
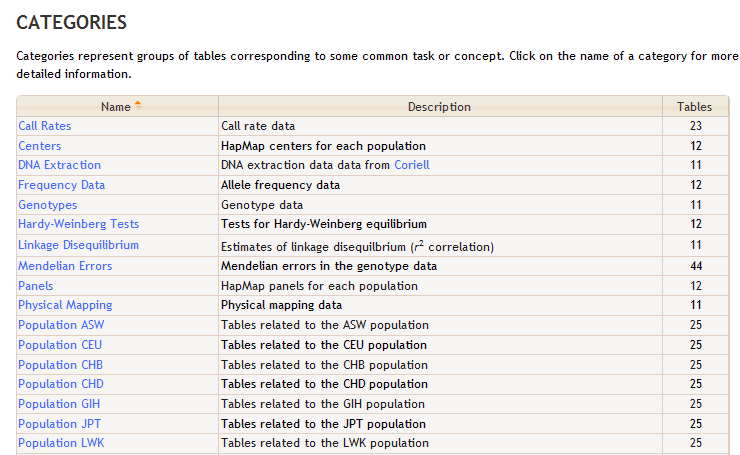
Child elements of <tags>
Element | Description | Notes |
|---|---|---|
<tag> | A tag and its tables |
|
<short_description> | Short description of the tag |
|
<long_description> | Long description of the tag |
|
<tables> | Tables used with the tag |
|
The <tag> element
This specifies a tag and its tables.
Attributes
Attribute | Value | Description |
tag_name | text | The label used in BioQ |
repeat | text | A space-separated list of values that substitute for $r in the other XML elements and attributes |
Tables: the <tables> element
Specify the tables in database. This populated the tbl table in the dbDoc relational database.
Example
<documentation>
<tables>
<table table_name="/^(\w{3})_snp_summary$/">
<short_descrip>A summary of SNP data for the _uc_($t1) population</short_descrip>
<long_descrip>
<p>A summary of SNP data for the _uc_($t1) population, including chromosomal mapping,
proportion of missing genotypes, number of Mendelian errors, p-value from test Hardy-Weinberg equilibrium,
allele frequency data and the number of LD proxies.
</p>
</long_descrip>
<column column_name="chr">
<short_descrip>Chromosome</short_descrip>
<long_descrip>
<p>Chromosome from build 132 of the dbSNP database (_db_link(dbsnp_human_132)), which uses the
<a href="http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/data/index.shtml" target="_blank">GRCh37</a>
human reference genome. Only SNPs that map to
unique coordinates (chromosome and position) are included in our implementation of the HapMap database.
We re-mapped the original HapMap-format genotype files to this reference genome using data from dbSNP. This data
is taken from the table _tbl_link($t1_snps).
</p>
</long_descrip>
</column>
</table>
</tables>
</documentation>
This produces the following output in BioQ.
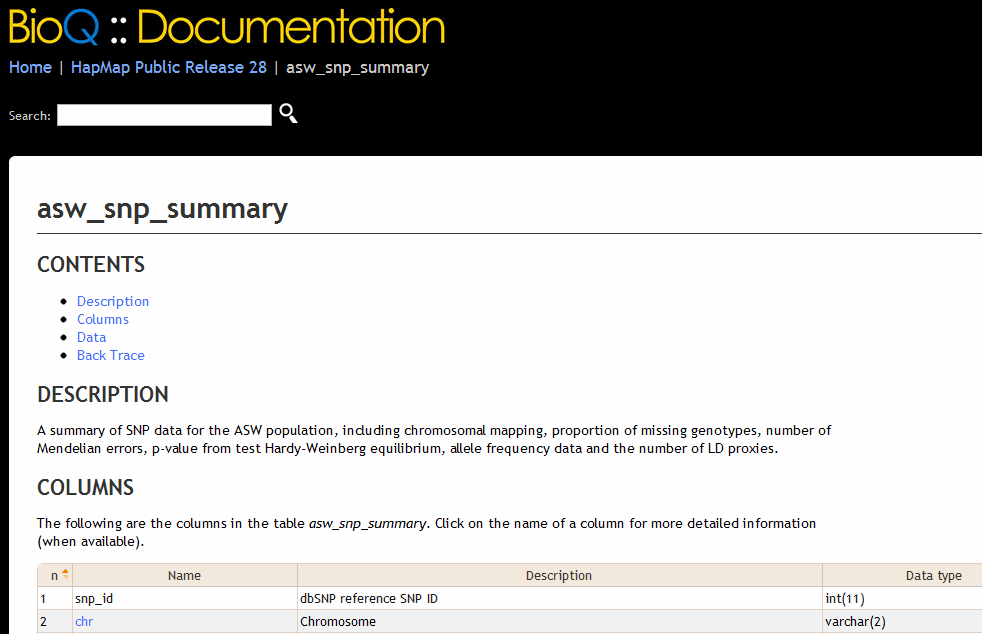
And after following the chr link we have the following page.

The <table> element
Specify a specific table within a <tables> element.
Attributes
Attribute | Value | Description |
table_name | text | The name of the table |
Regular expressions in the table_name attribute
When there are many tables in your database that all fit certain pattern, regular expressions can be used to simplify the documentation process.
table_name="/regular expression/"
The variable $t1 when then match the parentheses () used in the regular expression.
The <column> element
Specify a specific column within a <table> element. This populates the col table in the bioq_dbdoc relational database.
Attribute | Value | Description |
|---|---|---|
column_name | text | The name of the column |
Child elements of <column>
Element | Description | Notes |
|---|---|---|
<short_descrip> | Short description |
|
<long_descrip> | Long description |
|
Flow groups: the <flow_groups> element
Specify the flow groups in the BERT model for the current database. Flow groups are groups of tables that may be input or output for the various processes in the database.
Example
<documentation>
<flow_groups>
<flow_group group_name="genotypes" group_label="Genotypes">
<short_descrip>HapMap Genotypes</short_descrip>
<table table_name="/^(\w{3})_genotypes$/" filter="_uc_($t1)"></table>
<table table_name="/^(\w{3})_samples$/" table_type="subject" reference="true" filter="_uc_($t1)"></table>
<table table_name="downloads" reference="true"></table> <!-- *** TODO: show anything where filter IS NULL -->
</flow_group>
<flow_group group_name="call_rates" group_label="Call Rates">
<short_descrip>Genotyping call rates (determine proportion of missing genotypes)</short_descrip>
<table table_name="/^(\w{3})_plink_(Miss|imiss)$/" filter="_uc_($t1)"></table>
</flow_group>
</flow_groups>
</documentation>
This information is reflected in the flow diagram generated on this BioQ page. Here, the bold "S" over asw_samples indicates this table contains subjects (from the table_type attribute), and that this table and downloads are reference tables for this flow group due to the dashed lines.
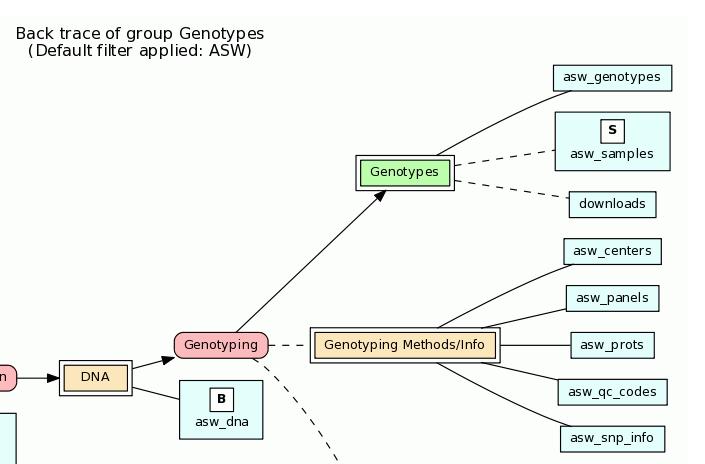
The <flow_group> element
Specify a single flow group.
Attribute | Value | Description |
|---|---|---|
group_name | text | The name of the group in the dbDoc database |
group_label | text | The label of the group |
Flow groups with no tables
This should be able to be done. However, each flow group may need to be output for some process.
Child elements of <flow_group>
Element | Description | Notes |
|---|---|---|
<short_descrip> | Short description |
|
<long_descrip> | Long description |
|
<table> | A table in the flow group | Regular expressions can be used |
The <table> element
Specifies a table in the flow group.
Attributes of <table>
Attribute | Value | Description |
|---|---|---|
table_name | text, | The name of the table. When a regular expression is used, $t1 will match what is in parentheses. |
table_type | biologic, | The type of the table in the BERT model. |
reference | true, | Whether this is a reference table for this flow group. |
filter | text | An optional filter for the table. |
Experimental processes: the <processes> element
A key component of BERT relational model are the experiments, and more generally the processes involved in generating the data in a genomic relational database. The flow groups described above can be input or output for a given process. The processes are entered into the <processes> section of the XML documentation.
Example
<documentation>
<processes>
<!-- Sample Collection -->
<process process_name="sample_collection" process_label="Sample Collection">
<short_descrip>The HapMap sample collection process</short_descrip>
<long_descrip>The HapMap sample collection process. While at the moment we do not
store detailed information on the HapMap collection process, this item serves as a placeholder
in our experimental process flow models.
</long_descrip>
<flow_group group_name="samples" flow_direction="output"></flow_group>
</process>
<!-- DNA Extraction From Coriell -->
<process process_name="dna_extraction" process_label="DNA Extraction">
<short_descrip>DNA extraction data from
<a href="http://ccr.coriell.org/" target="_blank">Coriell</a>
</short_descrip>
<flow_group group_name="samples" flow_direction="input"></flow_group>
<flow_group group_name="dna" flow_direction="output"></flow_group>
</process>
</processes>
</documentation>
This XML code leads to the follow documentation on the DNA Extraction process in BioQ.
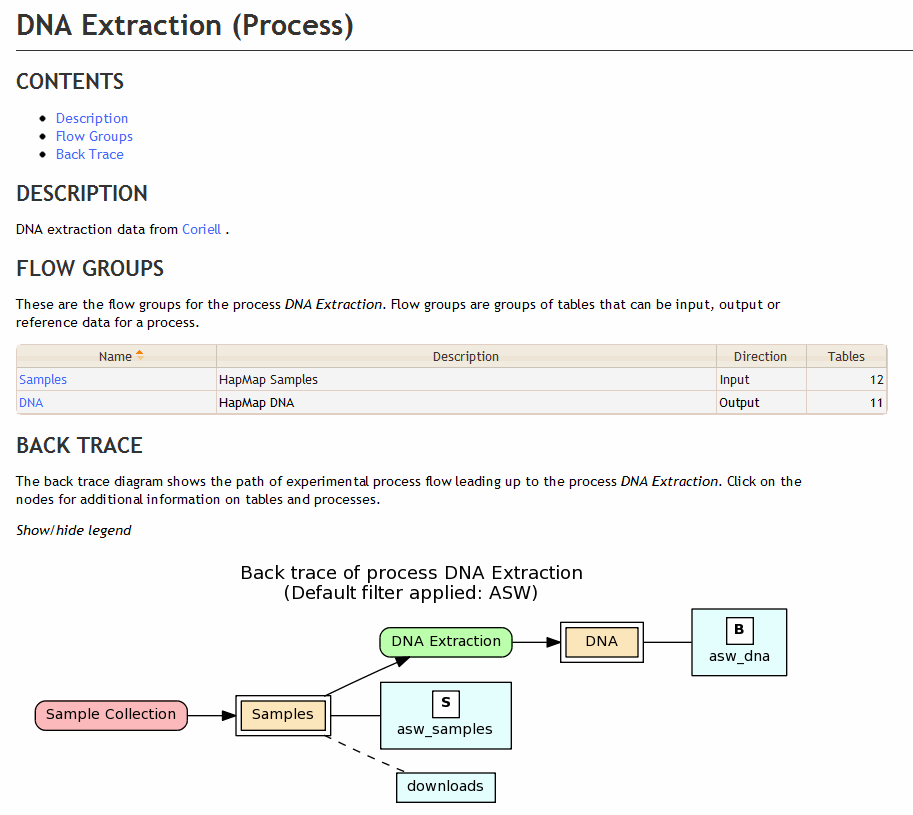
The <processes> element
Specifies a process in the BERT relational model.
Attributes of <process>
Attribute | Value | Description |
|---|---|---|
process_name | text | The name of the process in the bioq_dbdoc relational database |
process_label | text | The label of the process used in BioQ web pages |
experiment | true/false | Whether the process is an experiment, and therefore has at least one results table as output |
Child elements of <process>
Element | Description | Notes |
|---|---|---|
<short_descrip> | Short description |
|
<long_descrip> | Long description |
|
<flow_group> | Can be input, output or reference |
The <flow_group> child of <process>
A flow group for the process that can be either input, output or reference data for the process.
Attributes of <flow_group>
Attribute | Value | Description |
|---|---|---|
group_name | text | The name of the flow group in the bioq_dbdoc relational database |
flow_direction | input, | The direction of flow relative to the process |
Database queries: the <queries> element
The <queries> element contains the queries used in the BioQ::Query tools.
Example
The following example makes use of the repeat attribute of the <query> tab that allows queries to specified simultaneously for a number of tables. The variable $r takes on the value of the items in the list.
<query query_name= "$r_dna"
query_label= "_uc_($r) DNA"
order= "12"
doc_table= "$r_dna"
filter= "$r"
repeat= "asw ceu chb chd gih jpt lwk mex mkk tsi yri">
<short_descrip>_uc_($r) DNA extraction data from <a href="http://ccr.coriell.org/" target="_blank">Coriell</a>.</short_descrip>
<sql>SELECT QTBL.* FROM $db.$r_dna AS QTBL</sql>
<order_by>fam_id, ind_id</order_by>
</query>
This leads to the following BioQ query page and query results for the HapMap ASW population.
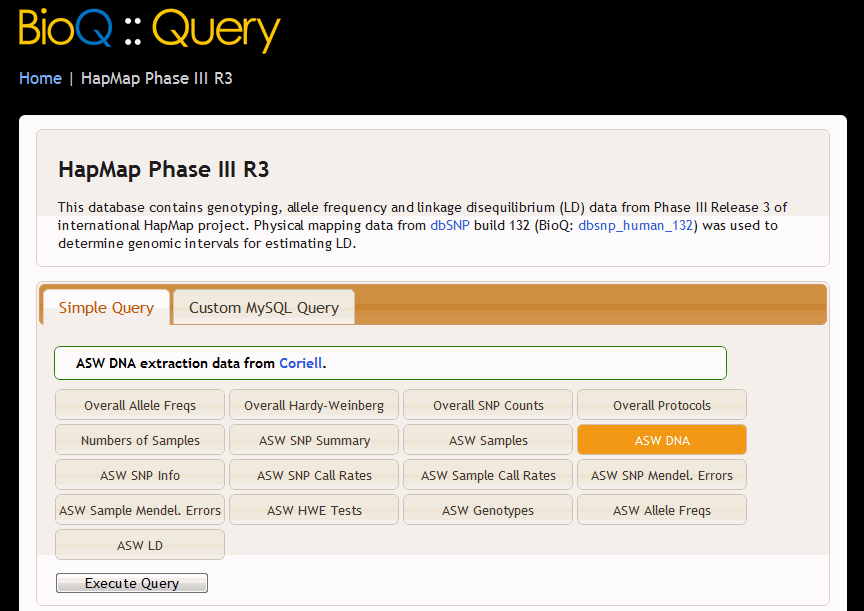
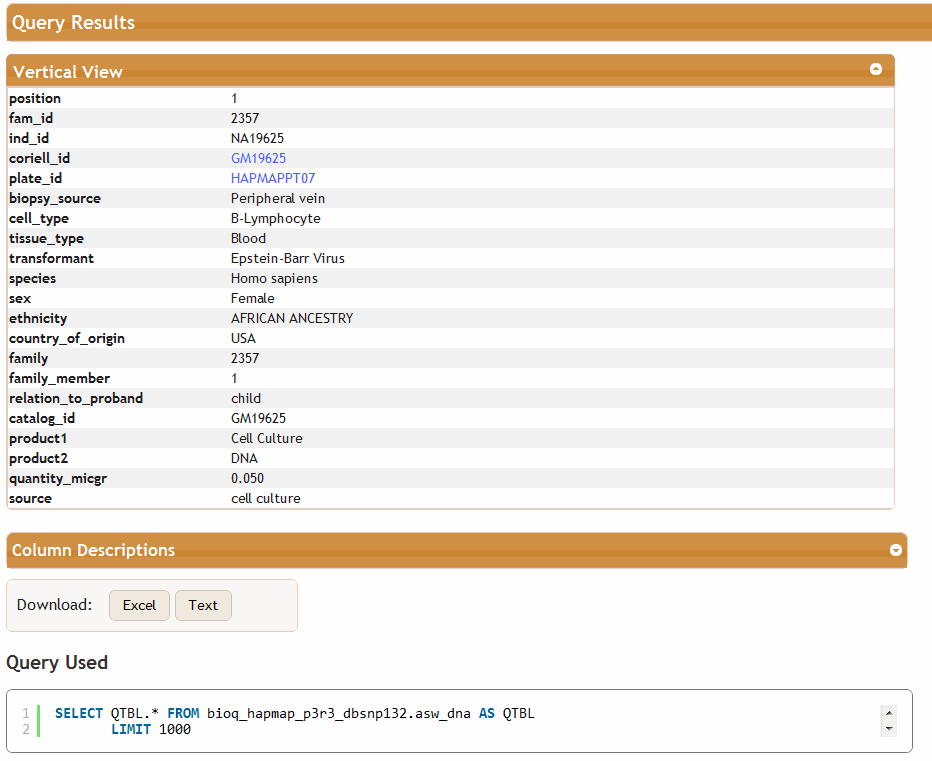
The <query> element
A specific query to be used in BioQ::Query.
Attributes of <query>
Attribute | Value | Description |
|---|---|---|
query_name | text | The name of the query in thebioq_dbdoc relational database. If the name has the form "_example_n" where n=1,2,3,..., then these queries will be used as examples in the advanced query section. |
query_label | text | The label used for the query in the BioQ::Query page |
order | integer | Used to specify the order of the queries in BioQ::Query |
doc_table | text | The table in the genomic database used for the Column Descriptions section of BioQ::Query |
filter | text | An optional filter that limits queries shown in BioQ::Query |
repeat | text | A space-separated list of values that substitute for $r in the other XML elements and attributes |
Child elements of <query>
Element | Description | Notes |
|---|---|---|
<short_descrip> | Short description |
|
<long_descrip> | Long description |
|
<sql> | SQL code used for the query | The expression "$db" will be substituted for the database being documented. Other expressions of the form %{text} will be substituted at run time by bioq_query.pl. Expression of the form ${var} according the variable assignments in the file specified in the --dbdoc-xml-var-file option to dbdoc_util.pl. |
<order_by> | SQL code used to sort the results | This will sort the results of the query rather than sort the entire source table prior to the query. If one actually includes an ORDER BY in the query code, then then entire source table will be sorted first. Do not use aliases here as shown in the example above. |
<where_cond> | The WHERE condition to use in the query. | Do NOT include the word "WHERE". It will be inserted. |
<column_map> | Indicates which columns in the query correspond to certain standard columns like snp_id. Example: snp_id:QTBL.snp_id (refers to alias QTBL). This is used, for example, in LD PROXY lookups. | Columns must be referred to using table names and/or aliases used in the query. This is a comma-separate list of <reference name>:<column name> values. The <column name> must include the alias used in the query, such as DBSNP.snp_id. |
Linking Functions
We have provided the following functions that insert hyperlinks to databases, tables and columns in the documentation.
Function | Description | Notes |
|---|---|---|
_db_link(database) | Link to database |
|
_tbl_link(table) | Link to table | The database must be clear from the context. |
_col_link(column) | Link to column | The database and table must be clear from the context |
_db_tbl_link(database.table) | Link to table |
|
_db_tbl_col_link(database.table.column) | Link to column |
|
Example
<documentation>
<tables>
<table table_name="/^(\w{3})_genotypes$/">
<short_descrip>Genotypes for the _uc_($t1) population</short_descrip>
<long_descrip>Genotypes for the _uc_($t1) population. The for each SNP the single column _col_link(genotypes) contains
all genotypes observed for the _uc_($t1) population at that SNP. The order of the genotypes corresponds to _tbl_column_link($t1_samples.position).
That is, the genotype at position N, N=1,2,3,..., corresponds
to the sample with _tbl_column_link($t1_samples.position) = N.
</long_descrip>
</table>
</tables>
</documentation>
This produces the following description in BioQ.
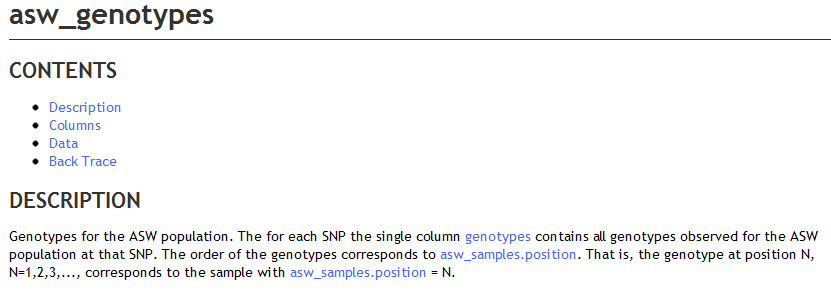
Text Functions
We have included some basic text functions such as uppercase.
Uppercase
_uc_(text)
Example
Convert the HapMap population names to uppercase.
<tag tag_name="Population _uc_($r)" repeat="asw ceu chb chd gih jpt lwk mex mkk tsi yri">
<short_description>Tables related to the _uc_($r) population</short_description>
<tables>/^$r_/</tables>
</tag>
Variable Substitution
- Expressions of the form ${name} are substituted according to values given the file provided in the --dbdoc-xml-var-file option to dbdoc_util.pl. These substitutions are made prior to storing the data in the dbdoc database.
- Expressions of the form %{name} are more complex
- Some will substituted by bioq_query.pl and must use the %special_vars names
- <sql> elements
- All others will be substituted by PHP and must use PHP constants
- Some will substituted by bioq_query.pl and must use the %special_vars names
In this example %{1kg_db} (note that there's no bioq prefix) is used by bioq_query.pl; the databases, tables and columns given are used to define interpopulation queries. The other expression %{bioq_1000genomes_db} is used by PHP to create the proper source table references.
<feature_table feat_table_name="feat_1kg_site"
label="1000 Genomes Sites"
primary_key="sites_id"
primary_key_data_type="INT"
interpop_position_type="position"
ref_pos_db="%{1kg_db}"
ref_pos_table="vcf_sites"
ref_pos_column="pos_global"
ref_pos_table_primary_key="n"
default_source_db="%{bioq_1000genomes_db}"
default_source_table="vcf_sites">
Examples of special variable substitution functions
- %latest_db() (implemented in CommonClass.php::substitue_special_vars()
Adding external columns
Sometimes a query will join data from external tables. In this case we may want to provide documentation for columns that are not actually in the table, but are in this external table. Here is an example from the 1000 Genomes queries:
<table table_name="/(\w{3})_vfreq/">
<short_descrip>Allele frequencies in the _uc_($t1) population</short_descrip
...
<column column_name="EXTERNAL:DP">
<short_descrip>Total Depth (from the table _tbl_link(vcf_sites))</short_descrip>
</column>
</table>
The method is to prefix the name of the column with "EXTERNAL:". This will insert documentation into the tbl table.
Procedure for adding new XML elements and attributes
- Modify the bioq_dbdoc database schema file using MySQL Workbench to incorporate the new element/attribute
- Export the schema to db_doc_model.sql: see The Documentation Database#Updatingthedocumentationschema
- Run update_schema.sh to incorporate the changes to the schema
- Edit bioq/perl/dbdoc/Process.pm::processXPath() to allow the new element/attribute to be processed from an XML.
- In some cases elements and attributes are processed automatically, and only require the schema to be modified. That is, any element or attribute found in the XML code is added to the database.
- Run dbdoc_util.pl updatedoc to process the XML file and add the new element/attribute to the bioq_dbdoc database
- Run commit.pl to dump the revised schema and commit it to Subversion
Strategies for writing XML documentation
Core and release-specific documentation
Because a database will often have several different versions and releases, it may be useful to have a "core" XML file with detailed documentation, and a separate release-specific file that has only the <db> element. The HapMap databases use this strategy.
Navigating the database
At the moment documentation must be written in XML. While future versions of our software may a include a graphical interface for entering documentation, the current system of writing the XML file is best accompanied by a program that can graphically browser your database. There are many clients for browsing a database. Our tool of choice is DbVisualizer. The screenshot below shows how the HapMap database can be navigated and the data browsed which can be helpful when writing the XML documentation.
Procedure for adding and updating documentation
Adding documentation for a new database
- First run dbdoc_util.pl with the initdoc command. This will create default documentation entries using the names of database, tables and columns found in the genomic relational databases themselves. Basically this creates a template in the documentation database that can be expanded with additional documentation from an XML file.
- Then run dbdoc_util.pl with the updatedoc command. This requires the option dbdoc-xml-file, the XML file that contains the documentation.
- We recommend this system of initializing and reading all the documentation from XML whenever changes to the documentation are implemented. Future implementations of this software may provide better tools for updating documentation, such as graphical interfaces and an XML export feature.
Example
The following shell script will initialize the documentation for the database bioq_hapmap_p3r3_dbsnp132 and then read the complete documentation from the files bioq_hapmap_core.xml (general documentation for all versions of the HapMap databases) and bioq_hapmap_p3r3_dbsnp132.xml (release-specific information that deals only with the <db> element).
#!/bin/bash
util_dir=/projects/bioinf/ssaccone/dbdoc_util
util_cmd=$util_dir/dbdoc_util.pl
export PERL5LIB=$util_dir
script_name=bioq_hapmap_p3r3_dbsnp132
database=bioq_hapmap # Used for 'Core' documentation XML file name
version=p3r3_dbsnp132 # Used for documentation specific to this version
#
# Initialize and Load Everything: ${database}_core + ${database}_${version}.xml
#
$util_cmd initdoc --options-file=$script_name.opt >$script_name.out 2>$script_name.err
$util_cmd updatedoc --options-file=$script_name.opt --dbdoc-xml-file=${database}_core.xml >>$script_name.out 2>>$script_name.err
$util_cmd updatedoc --options-file=$script_name.opt --dbdoc-xml-file=${database}_${version}.xml >>$script_name.out 2>>$script_name.err
# dbdoc_util.pl options used by script bioq_hapmap_pr28_dbsnp132.sh dbdoc-db= bioq_dbdoc_1 dbdoc-target-db= bioq_hapmap_pr28_dbsnp132 process-dir=. delete-temp-files= 1 verbose= 1 db-delete-temp-tables= 1 print-options= 1 db-host= localhost db-user= bioq db-password-file= ./passwd db-dump-dir=. ./dumps